
Biohub Releases Protein Biology World Model to Address Disease
Biohub, the non-profit research organization co-founded by Priscilla Chan, MD, and Mark Zuckerberg, has now unveiled the latest update to the ESM protein language model family, with expanded capabilities in binder design and protein function mapping for therapeutic discovery. The release comes just seven months after Biohub recruited the team behind EvolutionaryScale.
The system includes ESMC (Evolutionary Scale Modeling Cambrian), a language model trained on approximately 2.8 billion sequences drawn from a breadth of life, including organisms adapted to extreme environments, and more than 20,000 types of proteins found in the human body. Evolutionary information encoded in ESMC is translated into atomic-resolution protein structures and interactions using the design engine and prediction model, ESMFold2.
Alex Rives, PhD, head of science at Biohub and former chief scientist at EvolutionaryScale, presented the work at this week’s “AI in Biology” symposium at Cold Spring Harbor Laboratory.
These models aim to transform the earliest stages of drug discovery by making biology more programmable. While traditional discovery workflows rely on slow and resource intensive experimental screens to identify promising drug candidates, rational protein design guided by in silico predictions has the potential to dramatically accelerate development timelines.
“We’re at an exciting point in protein biology where accurate digital representations allow asking experimental questions at a scale that wouldn’t be possible in the laboratory,” Rives told GEN Edge.
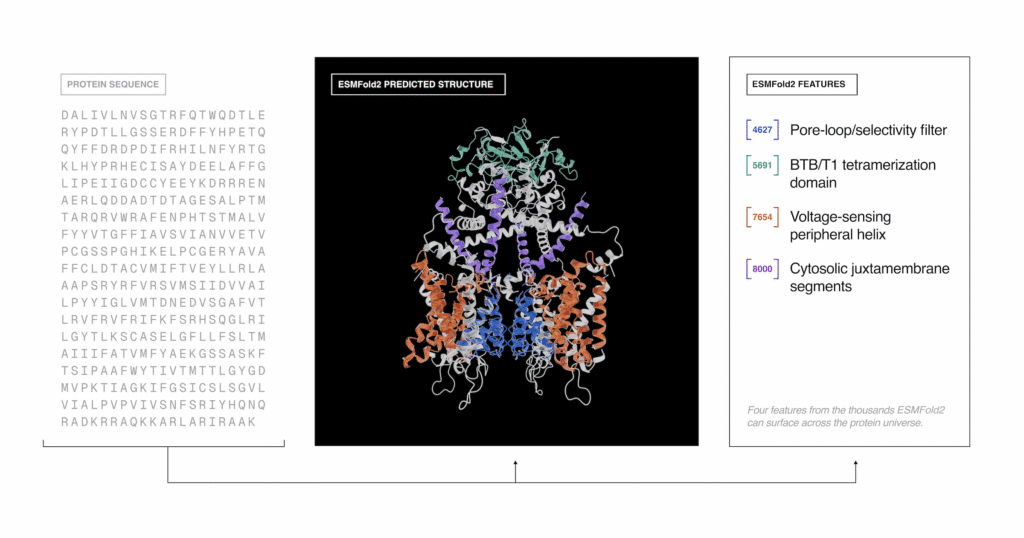
ESMFold2 designed high-affinity protein binders against five disease targets in cancer and immunology: receptor tyrosine kinases implicated in tumor growth (EGFR and PDGFRβ), immune checkpoints exploited by cancer cells to evade immune surveillance (PD-L1 and CTLA-4), and a regulator of immune cell signaling (CD45).
Lab-validated designs achieved hit rates ranging from 36–88% for compact mini-binders and 15–29% for antibody-derived formats, while also demonstrating nanomolar binding affinity, high specificity, and favorable stability profiles consistent with potential clinical utility. Notably, binders for PD-L1 showed therapeutic function and restored T-cell signaling in laboratory tests by blocking the same pathway as approved checkpoint therapies.
Rather than requiring multiple sequence alignments (MSAs) to build representations, ESMFold2 captures evolutionary information encoded during pretraining. The model also uses a looped transformer architecture, which allows compute to scale at inference time and avoids overfitting that can arise when training is constrained by limited experimental protein structures.
In benchmarking, ESMFold2 performed favorably when compared against Chai-1 from Chai Discovery, Boltz-1 from MIT (whose developers have since launched a public benefit corporation), and AlphaFold 3 from Google DeepMind.
The models are accessible under the highly permissive Massachusetts Institute of Technology (MIT) license for both commercial and non-commercial use. The work is described as a preprint that has not yet been peer reviewed.
“All in” on AI biology
Last November, Chan and Zuckerberg made the pledge to go “all in on AI-powered biology,” announcing that the organization’s scientific teams will now unite under a single entity, known as Biohub, where the duo would place the majority of their philanthropic effort.
Concurrently, Rives and EvolutionaryScale colleagues were recruited to tackle disease by decoding the “grammar” of amino acids through billions of years of evolution. That same mission had once secured a whopping $142 million seed round when the startup unveiled in 2024. The raise was led by Nat Friedman, Daniel Gross and Lux Capital, and included participation from Amazon Web Services (AWS) and NVentures, Nvidia’s corporate venture arm.
Biohub continues to advance virtual biology by building digital representations of molecules, genomes, cells, and living systems. The new ESM release joins Biohub’s growing ecosystem of biology models, including TranscriptFormer, which was published in Science earlier this month.
The organization recently invested $500 million in the Virtual Biology Initiative, a five-year campaign to accelerate the creation of technologies and multi-modal datasets to build predictive models of biology. The commitment comes a few months after the organization announced a collaboration with Arc Institute and Tahoe Therapeutics to build the largest single cell chemical perturbation dataset to power the virtual cell.
Evolution is all you need
Biohub has applied the new ESM models to generate the ESM Atlas, a mapping of 6.8 billion sequences and 1.1 billion predicted structures to protein function using ESMC’s representations. Generating this atlas would have taken “billions of years of experimental work,” but was condensed into a couple of weeks with computational inference. The ESM Atlas is released open-source.
By probing ESMC’s representations using sparse autoencoders (SAEs), a technique for identifying interpretable structure in large language models, the authors found that the model independently learned hierarchical organization covering the basic chemistry of individual amino acids, local structural interactions, and functional concepts across unrelated proteins, despite being trained only on sequence data.
Notably, ESM Atlas SAE feature clusters brought together RNA-guided DNA endonucleases, eukaryotic Fanzor proteins and their evolutionary ancestor, prokaryotic TnpB, despite their high evolutionary divergence and low sequence similarity. These insights could support the development of new gene-editing tools.
While the preprint’s results are still a step away from clinical impact, Rives reiterates the power of open science in placing these tools in the hands of researchers working directly in translational research.
Biohub is partnering with a number of platform partners, including AWS Bio Discovery, Benchling, Phylo, Tamarind Bio, Modal, Tool Universe, and SandboxAQ, to make the models widely available.
