
原标题:2-氨基-5-氯-N,3-二甲基苯甲酰胺的合成工艺
在农业生产中,害虫防治始终是保障粮食安全的重要课题之一。本世纪初,美国杜邦公司成功开发出具有创新结构的邻甲酰氨基苯甲酰胺类化合物——氯虫苯甲酰胺(chlorantraniliprole)(见图1)。该化合物通过激活昆虫鱼尼丁受体引发钙离子失衡,对鳞翅目、鞘翅目等多种害虫展现出卓越防效。相较于传统杀虫剂,其突出优势体现在高效低毒特性、优异的内吸传导性能以及延长持效期等方面,在玉米螟、甜菜夜蛾等重大害虫防控中具有重要应用价值。近年来随着氯虫苯甲酰胺专利保护期届满及国内应用需求持续扩大,2023年我国氯虫苯甲酰胺市场规模达到25亿元,我国科研机构在剂型研发与生产工艺方面取得突破性进展。据国家农业农村部农药检定所发布数据显示,截至2025年5月,氯虫苯甲酰胺产品登记达到570个,颗粒剂、水分散粒剂、悬浮剂、种子处理悬浮剂、超低容量液剂等为其主要登记剂型。
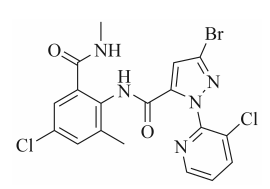
图1 氯虫苯甲酰胺结构式
在氯虫苯甲酰胺的合成工艺中,2-氨基-5-氯-N,3-二甲基苯甲酰胺作为关键中间体,其制备工艺的优化直接影响到终产品的成本效益与产业化可行性。目前该中间体的合成路线研究基础较为薄弱,尤其在原子经济性、催化剂选择及纯化工艺等核心环节缺乏系统性研究。据文献报道,合成2-氨基-5-氯-N,3-二甲基苯甲酰胺的方法主要有以下几种(见图2):1)以7-甲基靛红为原料,经氯化亚砜氯化、甲酸-双氧水氧化、甲胺化反应制备2-氨基-5-氯-N,3-二甲基苯甲酰胺,优化工艺条件下,反应总收率为62%;2)以2-硝基-3-甲基苯甲酸为原料,经加氢、环合、胺解和氯化反应得到2-氨基-5-氯-N,3-二甲基苯甲酰胺,反应总收率为77.6%;3)以2-硝基-3-甲基苯甲酸为起始原料,经甲酯化、铁粉还原、氯化亚砜氯化和甲胺氨解反应得到2-氨基-5-氯-N,3-二甲基苯甲酰胺,反应总收率为59.0%。
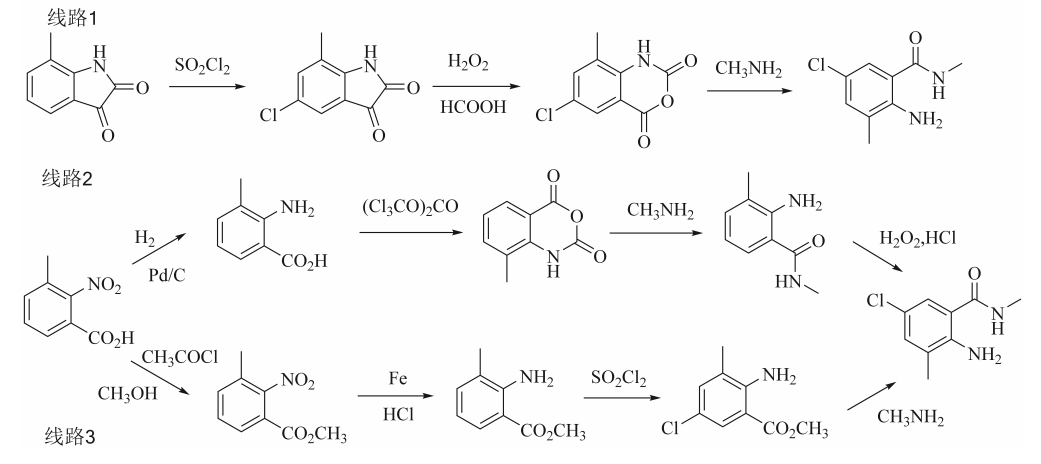
图2 2-氨基-5-氯-N,3-二甲基苯甲酰胺的合成路线
路线1虽然反应步骤短,收率相对较高,但是其起始原料7-甲基靛红价格较高,生产工艺成本高。合成路线2、合成路线3起始原料价格低,其中路线2反应收率也相对较高,但是由于起始原料2-硝基-3-甲基苯甲酸涉及安全问题,存在爆炸隐患,且路线3还涉及铁粉还原,因此这2条工艺路线不适合工业化生产。
在系统评估现有合成路径的生产效率与经济性基础上,本实验紧密围绕我国氯虫苯甲酰胺产业规模化发展的迫切需求,致力于开发具有完全自主知识产权的高效合成技术体系。本工艺以邻甲苯胺为起始原料,经盐酸-双氧水氯代、酰胺化-分子内Friedel-Crafts酰基化、水解开环、酰胺化4步反应合成2-氨基-5-氯-N,3-二甲基苯甲酰胺,通过优化工艺参数,解决了传统生产中废弃物处理成本高昂、环保达标难度大的行业痛点。新工艺采用价格低廉的邻甲苯胺作为起始原料,总原料成本大幅度降低,反应条件温和,三废排放量显著减少,更适合规模化制备生产。
1 实验部分
1.1 试剂与仪器
主要试剂:邻甲苯胺、草酰氯、氯化亚砜,化学纯,上海麦克林生化科技有限公司;三氯化铝、四氯化锡、多聚磷酸、三氯化铁、氯化锌、双氧水,分析纯,伊诺凯试剂公司;二氯甲烷、乙酸乙酯、无水硫酸钠、氢氧化钠,分析纯,国药集团化学试剂有限公司。
主要仪器:电子天平,AUY-120,日本岛津公司;低温恒温搅拌反应浴,DHJF-4002,郑州长城科工贸有限公司;电动搅拌器,JJ-1,江苏杰瑞尔电器有限公司;智能恒温数显水浴锅,HH-4,上海宏冠仪器;赛默飞液相色谱仪,UltiMate™ 3000,赛默飞世尔科技;Bruker 600MHz NMR型核磁共振仪,德国Bruker公司;IKA旋转蒸发仪,RV 8 V-C,艾卡(广州)仪器设备有限公司。
1.2 本文合成路线
新方法的工艺过程实现本质安全化设计,成功构建了可工程化放大的绿色合成路径,为产业化制备提供技术范式,具体合成路线见图3。
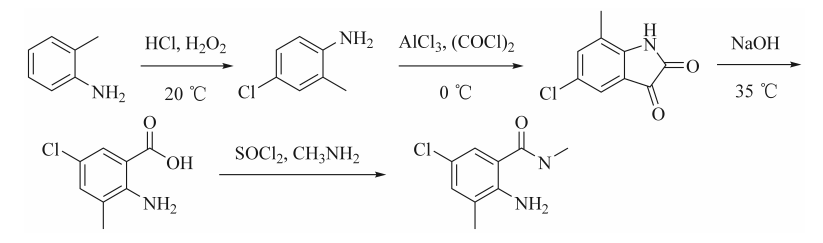
图3 本文2-氨基-5-氯-N,3-二甲基苯甲酰胺合成路线
1.3 化合物的合成
1.3.1 2-甲基-4-氯苯胺的合成
将100 g(0.93 mol)邻甲苯胺、680 g(2.79 mol)15%质量分数的盐酸、1 g氯化锌分别加入至2 L三口烧瓶中,水浴锅加热至20℃,开启搅拌,将320 g(1.41 mol)质量分数15%的双氧水通过高压液相泵加入反应瓶中,加毕后液相追踪监控邻甲苯胺剩余情况,反应约2.5 h后无邻甲苯胺剩余,加入33 g(0.48 mol)亚硝酸钠除去过量的双氧水,并向反应液中加入氢氧化钠调节反应液pH为7~8,然后将所有反应液倒入2 L萃取瓶中,加入400 g二氯甲烷分2次萃取反应液,合并所有有机相,加入30 g无水硫酸钠干燥有机相,过滤除去无水硫酸钠,有机相旋蒸脱除溶剂,得灰白色固体127 g(0.89 mol),收率为96.2%,液相纯度为99.2%,熔点为29.8℃。1H NMR(600 MHz,CDCl3)δ:6.98(d,1H),6.72(d,1H),6.47(dd,1H),3.61(s,2H),2.22(s,3H)。ESI-MS(m/z):[M+H]+142.1。
1.3.2 5-氯-7-甲基靛红的合成
将100 g(0.7 mol)2-甲基-4-氯苯胺、350 g二氯乙烷、85 g三乙胺(0.84 mol)、196 g(1.47 mol)三氯化铝加入1 L三口烧瓶中,冷阱温度设置为0℃,开启搅拌,将87.3 g(0.88 mol)草酰氯加入至恒压滴液漏斗中,缓慢滴加至反应液中,控制反应温度低于5℃,滴加完毕后升温至70℃继续反应,液相追踪反应进程,约1.5 h后检测无2-甲基-4-氯苯胺剩余,将反应液缓慢加入稀盐酸-冰水中淬灭,使用分液漏斗分出有机相,加入20 g无水硫酸钠干燥有机相,过滤除去无水硫酸钠,有机相旋蒸去除溶剂,得淡红色固体125 g(0.64 mol),收率为90.8%,液相纯度为99.5%,熔点为265.2℃。1H NMR(600 MHz,DMSO)δ:2.17(3H,s),7.22(1H,s),7.28(1H,s),11.14(1H,s,NH)。ESI-MS(m/z):[M+H]+ 196.1。
1.3.3 2-氨基-5-氯-3-甲基苯甲酸的合成
将100 g(0.51 mol)5-氯-7-甲基靛红、800 g(2 mol)质量分数为10%的氢氧化钠水溶液加入2 L三口烧瓶中,水浴锅升温至35℃,开启搅拌,将170 g(0.75 mol)质量分数为15%的双氧水通过恒压滴液漏斗滴入反应瓶,控制反应体系温度,液相追踪反应进程,反应约2 h,检测无5-氯-7-甲基靛红剩余,降温至25℃,用稀盐酸调节反应液的pH至4,过滤,所得滤饼用冰水洗涤,干燥滤饼,得淡黄色粉末85.5 g(0.46 mol),收率为90.2%,液相纯度为99%,熔点为196.2℃。1H NMR(600 MHz,DMSO)δ:7.61(d,1H),7.26(d,1H),2.16(s,3H)。ESI-MS(m/z):[M+H]+ 186。
1.3.4 2-氨基-5-氯-N,3-二甲基苯甲酰胺的合成
将80 g(0.43 mol)2-氨基-5-氯-3-甲基苯甲酸、80 g(0.67 mol)氯化亚砜加入至500 mL三口烧瓶中,油浴锅加热至80℃回流反应1.5 h,减压蒸馏去除反应液中的氯化亚砜,加入150 g二氯甲烷使反应液搅拌均匀,将反应液放置于冰水浴锅中,反应液温度在10℃以下,滴加50 g甲胺-二氯甲烷的混合液,其中甲胺16 g(0.51 mol),滴加过程保持反应温度低于20℃,滴加结束后继续反应1 h,反应完毕后,旋蒸脱除溶剂,得灰白色固体79 g,收率为92.5%,液相纯度为99.5%,熔点131.5℃()(文献值130~132℃)。1H NMR(600 MHz,DMSO)δ:2.14(s,3H),2.96(d,3H),5.6(s,1H),7.09(s,1H),7.16(s,1H)。ESI-MS(m/z):[M+H]+ 199.04。所得灰白色固体产品的核磁、熔点、质谱等结果与已确认结构的目标产品结果一致。
2 结果与讨论
2.1 2-甲基-4-氯苯胺的合成优化
使用双氧水-盐酸体系进行氯代反应,其中邻甲苯胺与双氧水的投料比例对反应收率有较大的影响,双氧水用量少会导致反应不完全,双氧水用量多会导致废水量的增加。在其他条件不变的情况下,考察双氧水用量的影响,邻甲苯胺与盐酸的摩尔比为1∶3,催化剂为氯化锌,用量为邻甲苯胺质量的1%,反应温度为20℃,液相追踪反应进程,结果如表1所示。
表1 双氧水用量对2-甲基-4-氯苯胺合成的影响

从表1可以看出,双氧水用量对2-甲基-4-氯苯胺合成有影响,其中邻甲苯胺与双氧水摩尔比为1∶1.5时收率为96.2%,继续提高双氧水用量对反应收率影响不大,同时双氧水用量过高也会导致废水量的增加,不利于产品的工业化生产。
相关氯虫苯甲酰胺文献中以氯化亚砜为氯化试剂、以乙腈为溶剂较为常见,文献最高收率为91.6%,且在原子经济性、三废产生量和原料成本等方面处于劣势。本实验路线氯正离子源为盐酸,与液氯、氯化亚砜等相比安全性更好,双氧水作为一种绿色的氧化剂,其使用比液氯、氯化亚砜等更加安全、方便,产生的氯正离子原位发生亲电取代,利用率高。
2.2 5-氯-7-甲基靛红的合成优化
2.2.1 催化剂种类对5-氯-7-甲基靛红合成的影响
以二氯乙烷为溶剂、三乙胺为缚酸剂,2-甲基-4-氯苯胺、三乙胺、催化剂、草酰氯的物质的量之比为1∶1.2∶2.5∶1.5,草酰氯滴加过程控制反应体系温度低于5℃,滴毕回流条件下考察催化剂种类对反应的影响,结果如表2所示。
表2 催化剂种类对5-氯-7-甲基靛红合成的影响

从表2可以看出,使用三氯化铝做催化剂制备5-氯-7-甲基靛红的收率最高,可以达到90.8%,选择以三氯化铝做催化剂进行后续反应。
2.2.2 催化剂用量对5-氯-7-甲基靛红的合成影响
以二氯乙烷为溶剂、三乙胺为缚酸剂、三氯化铝为催化剂,2-甲基-4-氯苯胺、三乙胺、草酰氯的物质的量之比为1∶1.2∶1.5,草酰氯滴加过程控制反应体系温度低于5℃,滴毕回流条件下考察催化剂用量对反应的影响,结果如表3所示。
表3 催化剂用量对5-氯-7-甲基靛红合成的影响

从表3可以看出,三氯化铝用量为2.1倍时反应收率最高,继续增加用量也无法进一步提高反应收率。
2.2.3 草酰氯用量对5-氯-7-甲基靛红合成的影响
以二氯乙烷为溶剂、三乙胺为缚酸剂,2-甲基-4-氯苯胺、三乙胺、三氯化铝的物质的量之比为1∶1.2∶2.1,草酰氯滴加过程控制反应体系温度低于5℃,滴毕回流条件下考察草酰氯用量对反应的影响,结果如表4所示。
表4 草酰氯用量对5-氯-7-甲基靛红合成的影响

从表4可以看出,当2-甲基-4-氯苯胺与草酰氯摩尔比为1∶1.25时反应收率最高,继续提高草酰氯投料对反应收率影响不大。
2.2.4 反应温度对5-氯-7-甲基靛红合成的影响
以二氯乙烷为溶剂,三乙胺为缚酸剂,三氯化铝为催化剂,2-甲基-4-氯苯胺、三乙胺、三氯化铝、草酰氯的物质的量之比为1∶1.2∶2.1∶1.25,草酰氯滴加过程控制反应体系温度低于5℃,考察滴毕草酰氯后反应温度对反应的影响,结果如表5所示。
表5 反应温度对5-氯-7-甲基靛红合成的影响
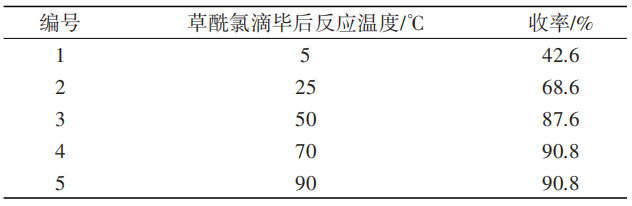
从表5中可以看出,滴加草酰氯结束后反应温度对收率有较大影响,其中70℃时反应收率最高。
2.3 2-氨基-5-氯-3-甲基苯甲酸的合成优化
双氧水浓度会影响5-氯-7-甲基靛红开环速率和反应收率,双氧水浓度过低会影响反应收率,双氧水浓度过高可能会存在安全隐患。在其他条件不变的情况下,5-氯-7-甲基靛红、氢氧化钠、双氧水物质的量比为1∶3.9∶1.47,反应温度35℃,考察双氧水浓度对反应的影响,实验结果如表6所示。
表6 双氧水浓度对2-氨基-5-氯-3-甲基苯甲酸合成的影响
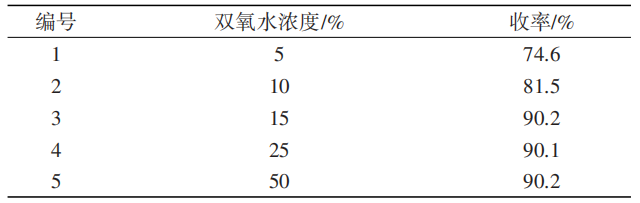
从表6可以看出,双氧水浓度为15%时收率最高为90.2%,继续提高双氧水浓度对反应影响不大,且双氧水浓度过高会存在安全隐患。
2.4 2-氨基-5-氯-N,3-二甲基苯甲酰胺的合成优化
在其他条件不变的情况下,2-氨基-5-氯-3-甲基苯甲酸与二氯亚砜摩尔比为1∶1.55,二氯甲烷为溶剂,反应温度低于20℃,滴毕后反应1 h,考察2-氨基-5-氯-3-甲基苯甲酸与甲胺的摩尔比对反应收率的影响,实验结果如表7所示。
表7 原料投料比对2-氨基-5-氯-N,3-二甲基苯甲酰胺合成的影响

从表7可以看出,2-氨基-5-氯-3-甲基苯甲酸与甲胺摩尔比为1∶1.2时收率最高,等摩尔投料反应收率不到70%,同时继续提高甲胺投料对反应收率影响不大。
3 结论
通过综合分析2-氨基-5-氯-N,3-二甲基苯甲酰胺合成工艺的基础上,开发了以邻甲苯胺为原料合成2-氨基-5-氯-N,3-二甲基苯甲酰胺新工艺,反应经盐酸-双氧水氯代、酰胺化-分子内Friedel-Crafts酰基化、水解开环、酰胺化4步合成产品,反应总收率72.8%(以邻甲苯胺计)。该工艺反应条件温和,原料易得,操作条件简单,适合工业化生产。